Laboratory Investigation (2020)
Stefaniya Boneva, Anja Schlecht, Daniel Böhringer, Hans Mittelviefhaus, Thomas Reinhard, Hansjürgen Agostini, Claudia Auw-Haedrich, Günther Schlunck, Julian Wolf #, Clemens Lange #
# contributed equally
Number of citations (crossref.org): Loading....
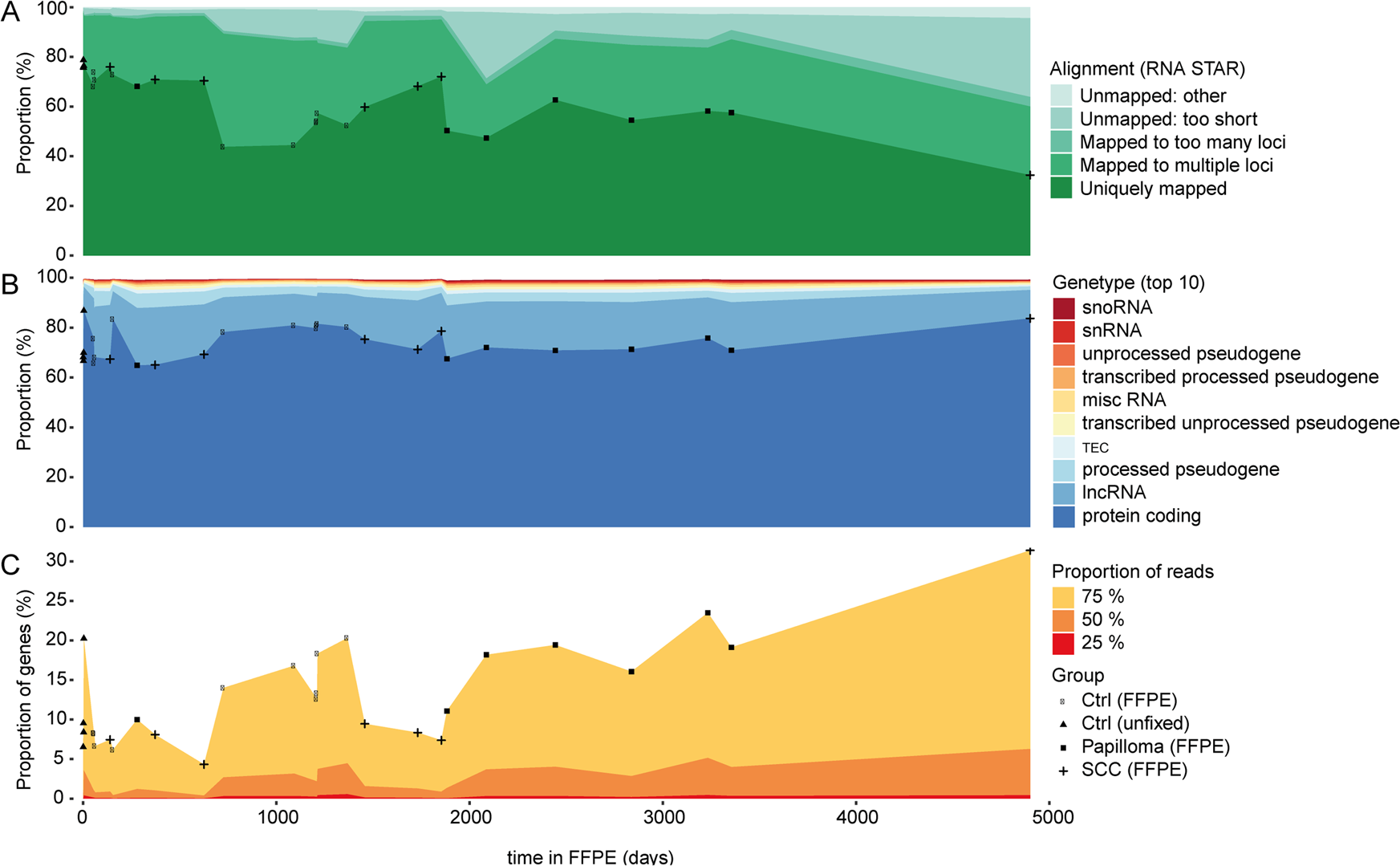
This study aims to compare the potential of standard RNA-sequencing (RNA-Seq) and 3′ massive analysis of c-DNA ends (MACE) RNA-sequencing for the analysis of fresh tissue and describes transcriptome profiling of formalin-fixed paraffin- embedded (FFPE) archival human samples by MACE. To compare MACE to standard RNA-Seq on fresh tissue, four healthy conjunctiva from four subjects were collected during vitreoretinal surgery, halved and immediately transferred to RNA lysis buffer without prior fixation and then processed for either standard RNA-Seq or MACE RNA-Seq analysis. To assess the impact of FFPE preparation on MACE, a third part was fixed in formalin and processed for paraffin embedding, and its transcriptional profile was compared with the unfixed specimens analyzed by MACE. To investigate the impact of FFPE storage time on MACE results, 24 FFPE-treated conjunctival samples from 24 patients were analyzed as well. Nineteen thousand six hundred fifty-nine transcribed genes were detected by both MACE and standard RNA-Seq on fresh tissue, while 3251 and 2213 transcripts were identified explicitly by MACE or RNA-Seq, respectively. Standard RNA-Seq tended to yield longer detected transcripts more often than MACE technology despite normalization, indicating that the MACE technology is less susceptible to a length bias. FFPE processing revealed negligible effects on MACE sequencing results. Several quality-control measurements showed that long-term storage in paraffin did not decrease the diversity of MACE libraries. We noted a nonlinear relation between storage time and the number of raw reads with an accelerated decrease within the first 1000 days in paraffin, while the numbers remained relatively stable in older samples. Interestingly, the number of transcribed genes detected was independent on FFPE storage time. RNA of sufficient quality and quantity can be extracted from FFPE samples to obtain comprehensive transcriptome profiling using MACE technology. We thus present MACE as a novel opportunity for utilizing FFPE samples stored in histological archives.